- Smoke Signals: Navigating the Evolving Landscape of Nicotine and Tobacco Regulations
- Posts
- June 24, 2024 - First menthol ENDS PMTAs authorized ⚖️
June 24, 2024 - First menthol ENDS PMTAs authorized ⚖️
Issue #12 - Deficiency Letter Deep Dive
Jon Dice
June 24, 2024
🔔 Reminder - A Tobacco Product Scientific Advisory Committee (TPSAC) meeting will be held on Wednesday June 26, 2024 at 9 am eastern to discuss the renewal of a risk modification order, submitted by Swedish Match USA, Inc. for several General Snus loose snus and portioned snus products. The meeting is open to the public.


CDISC, in partnership with the U.S. Food and Drug Administration Center for Tobacco Products (FDA CTP), is pleased to release the Tobacco Implementation Guide (TIG). The TIG is a Foundational Standard that serves as a comprehensive resource for the collection, tabulation, analysis, and exchange of tobacco product data for submissions to FDA CTP.
Use Cases Addressed in the TIG v1.0:
Product Description, which refers to concepts used to characterize tobacco products.
Nonclinical, which refers to concepts used to identify potential risks and effects on biological processes for tobacco products via in vitro and in vivo nonclinical studies.
Product Impact on Individual Health, which refers to concepts used to assess the impact of tobacco products on individuals.
Product Impact on Population Health, which refers to concepts used to assess the impact of tobacco products on populations of individuals.

The purpose of the ENDS Enforcement Task Force is to coordinate efforts and enhance enforcement against the illegal sale of unauthorized electronic nicotine delivery systems (ENDS) products. The Task Force brings together various government agencies, including the Food and Drug Administration (FDA), the Department of Justice's Consumer Protection Branch, the Bureau of Alcohol, Tobacco, Firearms and Explosives (ATF), the U.S. Marshals Service (USMS), the U.S. Postal Inspection Service (USPIS), and the Federal Trade Commission (FTC).
The Task Force aims to address the illegal importation, manufacture, distribution, and sale of ENDS products. It facilitates the sharing of market knowledge, enforcement experience, and operational abilities among the participating agencies. The Task Force regularly meets to discuss the actions being taken by each agency, identify and investigate entities engaged in illegal ENDS activities, determine appropriate enforcement authorities, and initiate actions under those authorities.
The Task Force considers a range of enforcement strategies, including facilitating the seizure of unlawful products, pursuing administrative actions leading to civil monetary penalties, developing actions to deter trafficking of ENDS products online and through the mail, initiating criminal prosecutions when appropriate, advancing injunctive actions to halt illegal distribution and sale, and suggesting legislative measures to combat the problem.
By coordinating efforts and sharing intelligence, the Task Force aims to develop innovative strategies to address the evolving problem of illegal ENDS products and protect consumers, especially youth, from the risks associated with these products.


Several stakeholders of the vaping industry participated in a hearing at the U.S. Senate Committee on the Judiciary - Combatting the Youth Vaping Epidemic by Enhancing Enforcement Against Illegal E-Cigarettes chaired by Senator Richard Durbin (D-IL).
Tony Abboud, Executive Director of the Vapor Technology Association, emphasized the dramatic decline in youth vaping rates in the United States, attributing it to a federal law raising the age to buy tobacco products to 21, which the Vapor Technology Association championed. The testimony also highlighted the need for marketing reforms and youth access restrictions to further reduce youth vaping while ensuring flavored vaping products remained available to adult smokers.
Brian A. King, PhD, MPH, Director of the Center for Tobacco Products at the Food and Drug Administration (FDA), who discussed FDA's premarket review of new products and its comprehensive actions to prevent youth use of e-cigarette products.
Arun G. Rao, Deputy Assistant Attorney General for the Consumer Protection Branch, U.S. Department of Justice, addressed the illegal sale of unauthorized electronic nicotine delivery systems (ENDS) products and the Department's commitment to using all available tools to protect Americans, especially youth, from the threat posed by unauthorized ENDS products.
David Spross, Executive Director of the National Association of Tobacco Outlets (NATO), expressed strong support for a well-functioning regulatory system and emphasized the need for enhanced enforcement against illicit vapor products to ensure compliance with FDA laws and regulations.
The speakers collectively addressed the decline in youth vaping rates, the need for marketing reforms and youth access restrictions, FDA's premarket review of new products, the illegal sale of unauthorized ENDS products, and the need for enhanced enforcement against illicit vapor products.
We seek authorization for FDA to collect user fees for e-cigarette manufacturers, all of whom are currently paying no fees to increase the current collections by 114 million dollars to account for the increased workload and to index all future collections for inflation.
Brian King, Director, Center for Tobacco Products U.S. Food and Drug Administration (link to transcript)


On June 21, 2024 the FDA issued marketing granted orders to NJOY LLC for four menthol-flavored e-cigarette products. These authorizations mark the first non-tobacco flavored e-cigarette products to be authorized by the FDA. NJOY LLC is an operating company of Altria. The time from submission to authorization was approximately 51 months. The tobacco-flavored versions of these products were authorized in April 2022 (NJOY ACE including the device) and June 2022 (NJOY DAILY).
NJOY ACE POD Menthol 2.4% (sealed, pre-filled, non-refillable pod)
NJOY ACE POD Menthol 5% (sealed, pre-filled, non-refillable pod)
NJOY DAILY Menthol 4.5% (disposable e-cigarette with a prefilled, non-refillable e-liquid reservoir)
NJOY DAILY EXTRA Menthol 6% (disposable e-cigarette with a prefilled, non-refillable e-liquid reservoir)
The authorization of the NJOY ACE Menthol appropriate for the protection of public health (APPH) designation was based on several key considerations, as outlined in the TPL document:
The PMTAs contained sufficient evidence to show that the new products have the potential to benefit adults who smoke combustible cigarettes (CC) and who switch completely or significantly reduce their CC use - p. 8.
The applicant proposed robust marketing plans that include restrictions beyond those required with PMTA authorization, such as limiting youth exposure to the new products by not engaging in social media promotions, using models over the age of 45 in marketing materials, and prohibiting the sale of NJOY ACE ENDS on third-party websites - p. 8.
The scientific review found that the new products' aerosols have fewer harmful and potentially harmful constituents (HPHCs) than CC mainstream smoke, and many of the HPHCs present in the aerosols have comparatively lower potencies than those present in CC smoke - p. 16.
The applicant submitted brand-specific data from an online, observational study that contained evidence to suggest that the menthol-flavored new products will promote switching or CC reduction among adults who smoke CC relative to tobacco-flavored ENDS - p. 37.
The TPL concluded that the PMTAs contain sufficient evidence demonstrating that the menthol-flavored new products have the potential to benefit adults who smoke CC, who switch completely or significantly reduce their CC use, that outweighs the risk to youth - p. 41.
At least two (new) supporting memoranda were relied upon in both the ACE and DAILY TPL reviews. Both of these should be FOIA’d to provide additional review clarity in the coming months.
• Genotoxicity Hazard Identification and Carcinogenicity Tiering of Constituents in ENDS Premarket Tobacco Product Applications (June 3, 2024)
• Calculating Excess Lifetime Cancer Risk in ENDS Premarket Tobacco Product Applications (June 3, 2024)
PMTA Timelines for Various MGO and MDOs
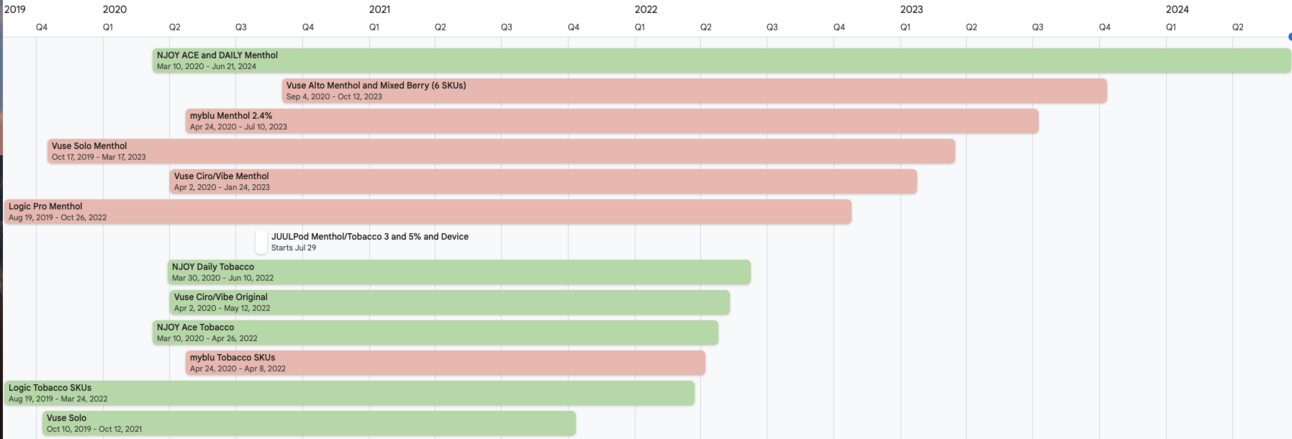
PMTA Timelines (click image for link)
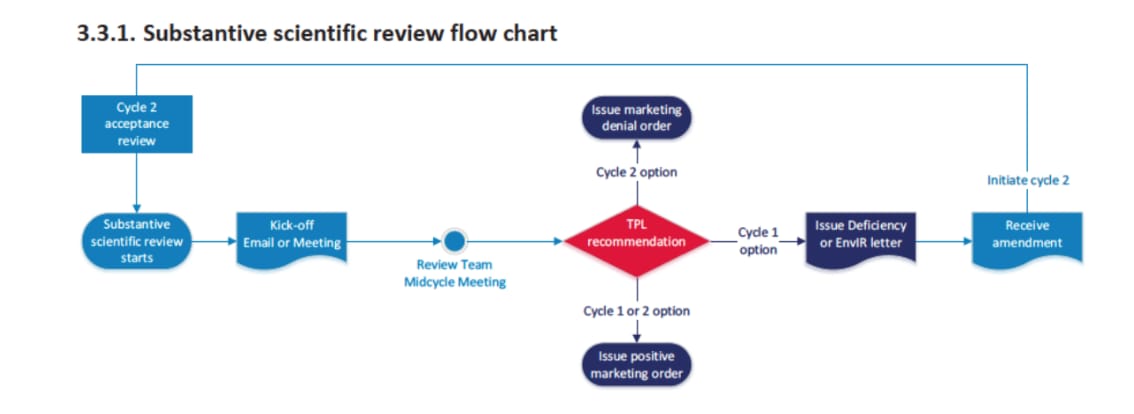
From PMTA Scientific Review Job Aid - Version 3/23/2021
Navigating the Deficiency Letter Response Process for PMTA and SE Applications
One of the critical steps in the Premarket Tobacco Product Application (PMTA) or a Substantial Equivalence (SE) application process is responding to a deficiency letter issued by the FDA. If you're fortunate enough to make it this far in the scientific review process there are some strategies to consider. This post will delve into the process of addressing a deficiency letter, highlight common scientific deficiencies, and provide strategies for effective responses. Additionally, the timelines associated with responding to these letters will be discussed.
Understanding the Deficiency Letter
A deficiency letter is issued by the FDA when an application lacks sufficient information or contains errors that prevent the agency from making a favorable determination. The letter outlines specific deficiencies that need to be addressed for the application to proceed. These deficiencies can range from missing data, inadequate study designs, to insufficient risk assessments.
Common Scientific Deficiencies
Incomplete or Inadequate Toxicological Data: One of the most frequent deficiencies is the lack of comprehensive toxicological data. This includes missing information on the toxicological profile of the product's ingredients, inadequate testing of harmful and potentially harmful constituents (HPHCs), and insufficient data on the product's impact on human health.
Insufficient Clinical and Non-Clinical Studies: Applications often fall short in providing robust clinical and non-clinical studies. This includes inadequate sample sizes, poorly designed studies, and lack of long-term data on product use and its health effects.
Inadequate Product Characterization: Another common deficiency is the lack of detailed product characterization. This includes incomplete information on the product's formulation, manufacturing process, and quality control measures.
Insufficient Behavioral and Perception Studies: Understanding how consumers perceive and use the product is crucial. Deficiencies in this area often include inadequate consumer perception studies, lack of data on product appeal, and insufficient information on potential misuse or abuse.
Inadequate Risk Assessment and Mitigation Strategies: Applications frequently lack comprehensive risk assessments and mitigation strategies. This includes insufficient data on the product's potential to reduce harm compared to existing products and inadequate plans to mitigate identified risks.
Strategies for Addressing Deficiencies
Thoroughly Review the Deficiency Letter: The first step in addressing a deficiency letter is to thoroughly review the document. Understand each deficiency in detail and prioritize them based on their impact on the overall application. This is also where any clarifying questions from the deficiencies would be surfaced.
Gather Comprehensive Data: Addressing deficiencies often requires gathering additional data. This may involve conducting new studies, reanalyzing existing data, or obtaining missing information from suppliers or third-party laboratories.
Enhance Study Designs: For deficiencies related to clinical and non-clinical studies, consider enhancing study designs. This may involve increasing sample sizes, extending study durations, and employing more rigorous methodologies.
Improve Product Characterization: Ensure that the product characterization is comprehensive. Provide detailed information on the product's formulation, manufacturing process, and quality control measures. Include data on batch-to-batch consistency and stability.
Conduct Robust Behavioral and Perception Studies: Address deficiencies in behavioral and perception studies by conducting robust research. Use validated survey instruments, ensure diverse and representative sample populations, and provide detailed analyses of the findings.
Develop Comprehensive Risk Assessments: For deficiencies related to risk assessments, develop comprehensive evaluations of the product's potential health impacts. Include comparative analyses with existing products and outline clear risk mitigation strategies.
Consult with Experts: Consider consulting with experts in toxicology, clinical research, behavioral science, and regulatory affairs. Their insights can help in addressing complex deficiencies and ensuring that the responses are scientifically sound.
Prepare a Detailed Response Document: Compile all the gathered data and analyses into a detailed response document. Address each deficiency separately, providing clear and concise explanations, supporting data, and references to relevant studies.
Timelines for Deficiency Letter Responses
The FDA typically provides a specific timeframe for responding to a deficiency letter. This timeframe can vary depending on the complexity of the deficiencies and the type of application. Here are some general guidelines:
Initial Review Period: The FDA aims to complete the initial review of a PMTA or SE application within 180 days. If deficiencies are identified, a deficiency letter is issued.
Response Timeframe: Applicants are generally given 90 days to respond to a deficiency letter. However, this period can vary based on the nature and number of deficiencies. In some cases, the FDA may grant extensions, but this is not guaranteed.
Review of Responses: Once the response to the deficiency letter is submitted, the FDA will review the additional information. This review period can take several months, depending on the complexity of the responses and the workload of the FDA.
Subsequent Deficiency Letters: It is possible to receive subsequent deficiency letters if the FDA determines that the responses are still inadequate. Each subsequent letter will have its own response timeframe, typically 90 days.
Resource Links
Chatbots
PMTA MGO Chatbot - This large language model (LLM) response agent was trained on several PMTA marketing granted orders and their respective technical project lead memos issued by FDA CTP. June 24, 2024 - the LLM was updated to include the NJOY ACE Menthol and DAILY Menthol MGOs and TPL review documents. This is very much a proof-of-concept exercise but responses are improving. Anthropic released Claude 3.5 Sonnet AI model on June 20, 2024.
I am making available a strategic analysis of CTP Application Job Aids. This resource provides a step-by-step analysis of the job aids provided to FDA CTP reviewers assigned to premarket tobacco product applications (PTMA), exemption request (EX) and substantial equivalence (SE) applications submitted under sections 910(a), 905(j)(A)(3) or 905(j), respectively, of the Federal Food, Drug, and Cosmetic Act (FD&C Act). The document includes the strategic analysis as well as the job aids.